АТРОФИЯ МЫШЦ ПРИ ЗАБОЛЕВАНИЯХ НЕРВНОЙ СИСТЕМЫ
Спасибо нашим инвесторам из казино онлайн
АТРОФИЯ МЫШЦ ПРИ ЗАБОЛЕВАНИЯХ НЕРВНОЙ СИСТЕМЫ
. I. А. м. п р и параличах церебрального происхождения можно наблюдать при детских церебральных параличах, т. е. при детских гемиплегиях, диплегиях и т. п.; при этом обыкновенно встречается атрофия не только мышц, но и костей; А. м. подвергаются б. ч. многие мышцы конечностей; подобные А. м. рассматриваются как задержка развития; в спинном мозгу в таких случаях замечается уменьшение количества серого вещества спинного мозга. А. м.
607
АТРОФИЯ МЫШЦ ПРИ ЗАБОЛЕВАНИЯХ НЕРВНОЙ СИСТЕМЫ
60S

можно встретить и у взрослых гемиплеги-ков, б. ч. спустя некоторое время после появления гемиплегии; поражаются проксимальные отделы конечностей и притом больше—верхней конечности; иногда при этом наблюдается атрофия костей; а также вазо-моторно – трофические расстройства. Реакции перерождения, как правило, не бывает. Появление подобных А. м. объясняется недостаточной трофической регуляцией мышечной ткани со стороны серого вещества спинного мозга вследствие выпадения церебральных трофических центров.—II. А. м. при параличах спинномозгового происхождения. При всякого рода токсиинфекцион-ных процессах, поразивших передние рога спинного мозга (напр., при полиоми-элите), при различных деструктивных процессах в них (на-прим., при кровоизлиянии) можно! наблюдать прогрессивно развивающиеся А. м. (см. рис. 1). Данный вид А. м. характеризуется наличием фибриллярных подергиваний и изменением электровозбудимости — от количественного понижения до полной реакции перерождения; часто также симметричным типом расположения А. м. При детском остром полиомиэлите спустя нек-рое время в парализованных мышцах развивается А. м., захватывающая обыкновенно бблыную часть мышц конечности; со временем наступает задержка и в развитии костей данной конечности; появляются вазомоторно-трофические расстройства; в исходном стадии болезни А. остается строго локализованной в одной или нескольких мышцах одной конечности. При хрон. полиомиэлите взрослых также развивается А. м., аналогичная описанной. А. м. встречаются также при гематомиэлии, различных миэлитах, сирингомизлии, иногда при спинной сухотке (т. н. tabes amyotrophica).— III. А. м. при поражении периферических нервов, развивающиеся при поражении нервных сплетений, двигательных или смешанных нервов (см. рисунок 2), характеризуются появлением реакции перерождения, периферическим распределением и регрессивным течением при улучшении паралича; при этом б. ч. не наблюдается фибриллярных подергиваний. А. м. этого рода наблюдаются при полиневритах различного происхождения, напр., после тифа, вследствие злоупотребления алкоголем, отравления свинцом, мышьяком. А. м. развиваются в парализованных мышцах большей частью довольно быстро и бывают резче выражены на периферии конечностей, больше в разгибателях, чем сгибателях.—IV. А. м.
Рисунок 1. Атрофия мышц плечевого пояса правой стороны при poliomyelitis ant. т. н. функционального происхождения. Истерическая А. м. всегда ограничивается парализованной конечностью, захватывая большей частью несколько сегментов конечности; атрофия мышц появляется спустя нек-рое время после наступления паралича и быстро сглаживается по его исчезновении; реакции перерождения не наблюдается.
Лит.:
Капустин A. A., Amyotrophiae tabi-dorum, «Невролог. Вестник», т. XXI, вып. 4, 1914; М а р г у л и с М. с, Мышечная атрофия при церебральных поражениях, дисс, М., 1907; Корнилов А., Мышечные похудания при страданиях суставов, костей и прилежащих частей, т. I и II, М., 1895; Leri A., Atrophies musculaires; Marie P., La pratique neurologique, P., 1911. А. Капустяи.
Атрофия иышц Арап-Дюшена
(Aran-Du-chenne). Под таким названием в неврологической литературе долгое время описывалась болезнь, представляющая собой подвид прогрессивной мышечной атрофии, стоящий рядом с миопатией, но отличающийся от нее целым рядом клинич. признаков: 1) семейный момент, столь свойственный миопатии, здесь не имеет места, 2) болезнь начинается не в детстве, как миопа-тия, а в более позднем, б. ч. зрелом возрасте, чаще поражая мужчин, 3) начинается процесс с верхних конечностей, симметрично и постепенно распро-страняясь от концевых отделов к корню конечности, 4) А.— в противоположность миопати-ческим А.—не сопровождаются псевдогипертро-фией мышц, но сопровождаются фибрилляр ными подергиваниями и реакцией перерождения, что указывает на их миэлопатическое происхождение и чему соответствует 5) лежащая в основе болезни А. двигательных клеток передних рогов спинного мозга. Отличием от хрон. переднего полиомиэлита считалась симметричность процесса при А. м. А.-Д., а также то, что, в противоположность полиомиэлиту, А. не следуют за парезами или параличами мышц, а развиваются первично, при чем сила пораженных мышц страдает лишь пропорционально степени постепенного исчезновения самой мышечной ткани. Болезнь протекает, очень медленно прогрессируя, и начинается с А. мелких мышц кистей (обычно сначала на правой руке),
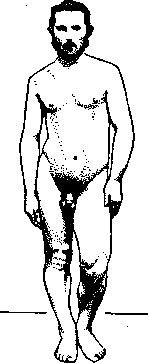
Рисунок 2. Атрофия мышц правой
ноги при заболевании п. ischia – dici. приводя к образованию т. н. «руки Аран-Дюшена»; медленно генерализуясь, А. могут, в конце-концов, захватить и бульбар-ную мускулатуру; сухожильные рефлексы исчезают; расстройства чувствительности, сфинктеров и интеллекта отсутствуют. В дальнейшем, большинство случаев. трактовавшихся как А. м. А.-Д., оказалось принадлежащим к другим болезненным формам: к сирингомиэлии, часть к боковому амиотро-фическому склерозу, к шейному гипертрофическому менингиту, к гипертрофическому невриту, к проф. амиотрофии, к туберкулезу позвоночника и пр. Различие между А. м. А.-Д. и хрон. передним полиомиэлитом также оказалось излишне схематизированным и искусственным (см.
Полиомиэлит).
Оставшиеся случаи, отвечающие клинич. типу Аран-Дюшена, подверглись дальнейшему расчленению, т. к. оказалось, что сифилис может давать сходные формы. Нек-рые авторы, особенно французские, вообще отрицают существование прогрессивной А. м. А.- Д. не-сифилитического происхождения. Другие, основываясь на единичных наблюдениях, продолжают описывать эту форму как самостоятельное заболевание, тождественное с хрон. передним полиомиэлитом (Dejerine) или даже независимое от него. Поэтому понятно, что теперь часто предпочитают пользоваться термином «А. м. А.-Д.» не для обозначения особой болезни, а лишь для обозначения особого клин, синдрома, отвечающего описанию Арана и Дюшена и наблюдаемого при разнообразных болезнях спинного мозга. Если эта – не-семейная социальная прогрессивная мышечная атрофия является, т. о., до настоящего времени спорной, то, наоборот, семейн. прогрессивная мышечная атрофия спинального происхождения оказалась твердо установленной.
Лит.:
Рот, Мышечная сухотка, М., 1895; А м м о-с о в, «Анн. Нерв. Клин. Бак. Ун-та», 1921; Маргу-л и с, Deutsche Zeitschr. f. Nervenheilk., В. LXXXVI. Атрофия мышц невральная, тип Шарко-Мари (син.: невротическая мышечная атрофия Гофмана, перонеальная атрофия Тут-са, периферический тип мышечной сухотки В. К. Рота, спинально-невритическая форма мышечной атрофии Бернгардта), описана в 1886 г. Шарко и Мари во Франции и Тутсом в Англии. Термин «невральная» амиотрофия предложен Гофманом. А. м. н. представляет наследственное заболевание, главным симптомом которого является прогрессирующая амиотрофия периферических отделов нижних и верхних конечностей. Процесс начинается постепенно, в возрасте до 20 лет (возможны большие возрастные отклонения). Сначала атрофируются мышцы стоп и голеней, а впоследствии—кистей и предплечий. Мышцы корня конечностей, Лица и туловища обычно не поражаются. Бедра худеют только в своей нижней трети, что придает им характерную коническую форму (см. рисунок 3). Часто—pes excavatus (но, в отличие от Фридрейховской стопы,—без гиперэкстензии основной фаланги большого пальца). Амиотрофия сопровождается потерей сухожильных рефлексов, реакцией перерождения, иногда—фибриллярными подергива – ниями; сухожильно-мышечные укорочения редки. Типичны расстройства чувствительности на дистальных отделах конечностей (гип – или анэстезии, иногда замедление болевой проводимости). Могут присоединяться парэстезии, стреляющие боли, вазомоторные расстройства, реже—атаксия, расстройство зрачковых рефлексов, спазмы, дрожание. Хождение почти всегда возможно, но походка извращена в форме т. н. steppage (петушиная походка). Стоя на одном месте, больные нередко вынуждены постоянно переминаться с ноги на ногу (т. н. pietinement). Болезнь поражает сначала ноги; лишь спустя несколько лет аналогичные явления замечаются и в руках. В дальнейшем—нередко стационарное течение. Болезнь наследственна и, как показали современные исследования, следует моногибридно-доминантному типу наследования. Мужчины заболевают в lVs раза чаще женщин. В некоторых из прослеженных в отношении данного заболевания семей А. м. н. передавалась в виде отклоняющихся от основного типа вариантов (например, с поздним началом или с атрофией зрительных нервов, или с участием только верхних конечностей, или с признаками гипертрофии периферич. нервных стволов). Па-толого – анатомически находят, большей частью, одновременное заболевание периферических нервов (в __jj виде дегенеративного» неврита) и спинного мозга (в виде склероза задних столбов и атрофии двигательных клеток передних рогов); могут быть поражены и другие системы (боковые столбы, столбы Кларка, межпозвоночные ганглии). Диференциаль-ный диагноз может быть труден лишь в отношении несколько сходной по своим симптомам миопатии дистального типа и гипер-трофическ. неврита. Спорадические с
лучаи невральной атрофпи мышц легко могут быть смешаны с полиневритом (в спорных случаях течение болезни уясняет диагноз). Прогноз при этой болезни плох, хотя кое-какая работоспособность и походка могут сохраняться долго. Причинное лечение болезни невозможно, но во многих случаях приносит пользу местное ортопедическое лечение. С профилактической точки зрения больным должен быть дан совет не иметь детей; здоровые же члены семьи лишь в совершенно исключительных случаях имеют шансы передать болезнь потомству^

Рисунок 3. Прогрессивная мышечная атрофия Шарко-Мари.
Лит.: Рот В. К., Мышечная сухотка, 1895; Kugelgen, Archiv f. Psychiatrie, В. XLV, 1909 (лит. вопроса); Давиденков С, Zeitschr. f. d. ges. Neurologie, В. CVII—CVIII; Lyon, Riv. di pat. nerv., 1926; Charcot et Marie, Revue de medecine, 1886; Hoffmann, Archiv f. Psychiatrie, 1889; Pette, Zeitschr. f. die ges. Neurologie, B. XCII. Атрофия мышц спинальная прогрессивная семейная, известна в двух формах: 1) детская и 2) сшшальная семейная амиотрофия взрослых. Последнее заболевание представляет собой наследственный хронический передний полиомиэлит (известно из очень небольшого числа наблюдений). В случаях, дошедших до аутопсии, анатомически определялся poliomyelitis anterior chronica; заболевание передавалось в семьях, невидимому, по доминантному типу. Лит.: Browning, W., Neurographs I, 1907; Dana, Journ. of nerv. a.ment. dis., 1914;Bruining, Deutsche Zeitschr. f. Nervenheilkunde. B. XXVII,1904. Атрофия прогрессивная мышц типа Вер-дниг-Гофмана, семейное заболевание, описанное впервые Werdnig’OM (1891 г.); спи-нальное происхождение болезни было установлено Hoffmann *ом, наблюдавшим в трех семьях 19 аналогичных случаев. Это— заболевание детского возраста, прогностически весьма неблагоприятное, клинически выражающееся в быстром развитии мышечных атрофии, распределяющихся довольно сходно с миопатическими, но зависящих от гибели двигательных клеток передних рогов спинного мозга. Болезнь начинается рано, обычно на 1-м году жизни, и спустя 3—6 лет приводит к смерти. А. развиваются постепенно. Раньше всего страдают, как и при миопатии, мышцы туловища, тазового пояса и бедер, в дальнейшем процесс распространяется повсюду. А. сопровождается потерей сухожильных рефлексов, реакцией перерождения, иногда фибриллярными подергиваниями. Псевдогипертрофия мышц отсутствует. Часто—гипотония мышц, иногда—вторичные контрактуры. Интеллект, зрачки, чувствительность, координация движений, сфинктеры — не страдают. Болезнь может быть и врожденной; в других случаях, наоборот, процесс начинается поздно, и смерть наступает только на втором десятилетии жизни. Иногда наблюдались сколиоз, бульварные симптомы; в нескольких более атипичных семьях наблюдали даже псевдогипертрофию икроножной мускулатуры. Генетика этого заболевания еще не ясна. Описаны и спорадические случаи; часто заболевают несколько братьев и сестер-—• детей здоровых родителей. В современной литературе форма А. п. м. типа В.-Г. трактуется обычно как рецессивная. Пат.-анатомически находят дегенерацию и гибель клеток передних рогов спинного мозга, с вторичной дегенерацией двигательных волокон в передних корешках и периферич. нервах, а также дегенеративные изменения в самих мышцах. К этому могут присоединяться перерождения и в других системах; наприм., дегенерация клеток Кларковых столбов, атрофические изменения в задних столбах спинного мозга. Диференциальный диагноз может представлять большие трудности в отношении миопатии. Прогноз абсолютно плох. Лечение невозможно. За последнее время стремились объединить атрсфию прог – рессивную мышц типа В.-Г. и т. н. myatonia congenita Оппенгейма (см. Миатония), — вопрос, остающийся открытым. Лит.: WerdDig, Archiv f. Psychiatr., B. XXII; Hoffmann, Deutsche Zeitschr. t. Nervenheilkunde, B. Ill, X u. XVII; Bruno, Neur. Zentr., 1906 u. Deutsche Zeitschr. f. Nerv., 1901. Атипические случаи см. у Emanuel, N. Zentr., 1912. С. Давиденков. Atrophia olivo-ponto-cerebellaris, см. Мозжечок. Artropbia olivo-rubro-cerebellaris, см. Мозжечок.